
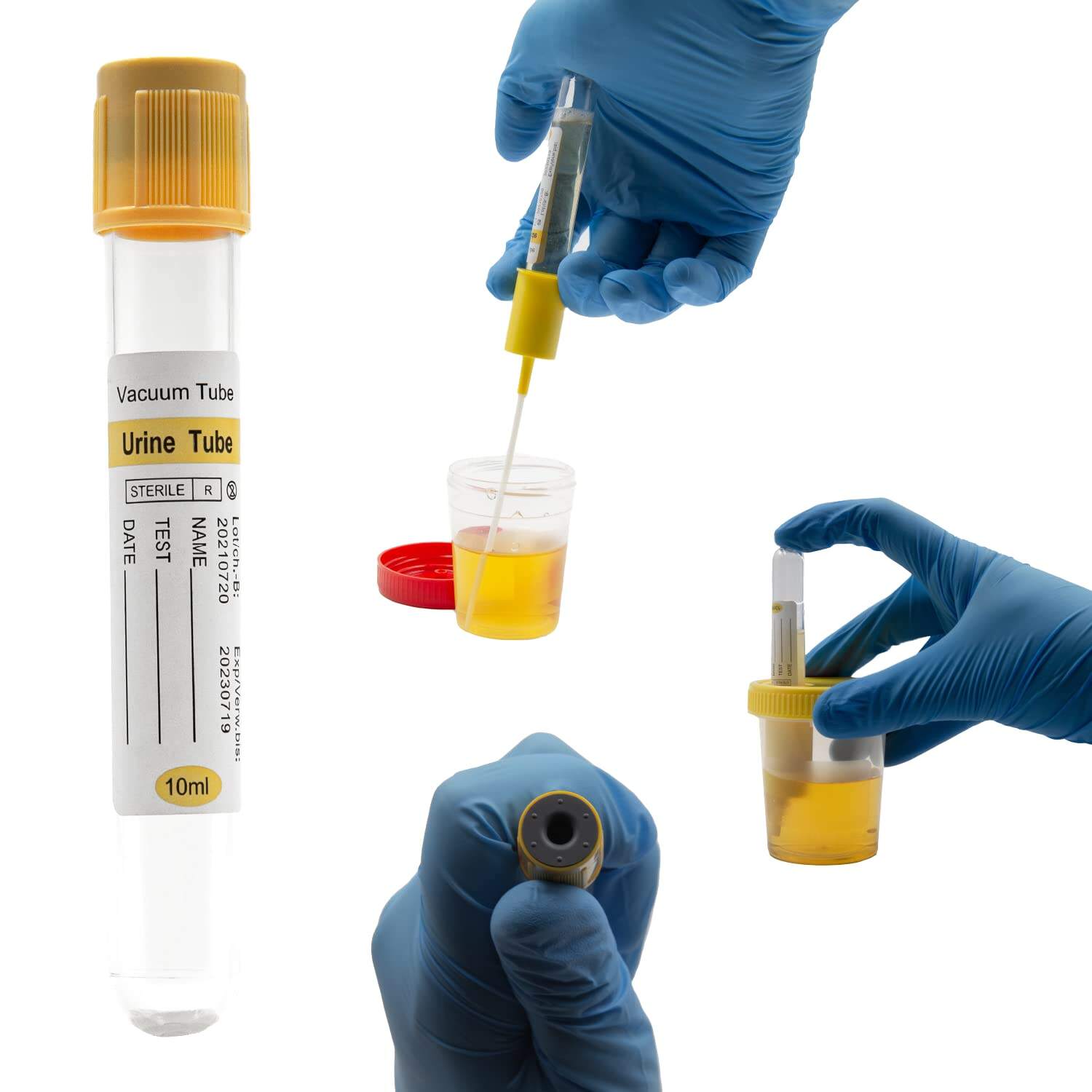
Understanding Regulatory Requirements for Vacuum Urine Containers
ISO 13485:2016 essentials: Design control, traceability, and sterile barrier system validation
ISO 13485:2016 establishes comprehensive quality management requirements for medical devices, including vacuum urine containers. The standard mandates formal design controls to ensure performance specifications align with user needs, supported by documented verification and validation. Risk management is integrated throughout development, often using tools like FMEA to proactively identify potential failures.
Good traceability systems need those unique IDs to follow parts all the way from when they first arrive at the factory until they become finished products. Without proper tracking, companies can't quickly find out where problems happened if something goes wrong later on. For medical devices especially, manufacturers have to test their sterile packaging under really tough conditions that mimic what might happen during shipping. These tests look at things like extreme temperatures and physical stress according to standards set by ISO 11607. The actual testing process checks whether germs can get through the packaging over time. Most companies run these tests once a year just to make sure nothing changes in the materials or manufacturing methods that could compromise sterility during storage before the product reaches patients.
FDA classification, 510(k) pathway, and UDI compliance for vacuum urine containers
Vacuum urine containers are classified as FDA Class II devices, requiring 510(k) clearance before market entry. Manufacturers must demonstrate substantial equivalence to a predicate device using comparative bench testing and biocompatibility data. The 510(k) submission includes performance, labeling, and sterilization details to support safety and effectiveness.
Under the Unique Device Identification or UDI rule, medical devices need to have their own special code that people can read as well as machines. These identifiers help track where devices go through the whole supply chain from manufacturer to end user. Companies are required to send all this UDI information into something called the Global Unique Device Identification Database, commonly known as GUDID. When problems occur after products hit the market, regulators can actually connect complaints or safety issues back to specific production runs thanks to this system. For quality management purposes, organizations also need to keep detailed records about customer complaints and make sure any fixes they implement get linked properly to those UDI codes. This creates better accountability for manufacturers while helping them stay on top of regulatory requirements.
Evaluating Manufacturer Compliance: Certification, Materials, and Sterility Assurance
Verifying ISO 13485 certification scope — does it explicitly cover vacuum urine containers?
Before purchasing vacuum urine collection systems, make sure the manufacturer holds an ISO 13485:2016 certificate that actually covers these specific products. Some certifications might list general medical equipment or just apply to office operations, which doesn't necessarily mean their factory meets standards for actual device manufacturing. Always ask for a copy of the complete certification paperwork and check directly with the regulatory authority that issued it. This extra step can save headaches down the road when ensuring proper quality control throughout the supply chain.
Generic certifications without specific product-line coverage contributed to 23% of FDA Form 483 observations in 2023 due to inadequate scope definition. Audit reports should confirm that the certification applies to the actual manufacturing lines producing vacuum urine containers, not just administrative functions.
Material safety: PVC-free alternatives, ISO 10993 biocompatibility, and extractables testing
Opt for PVC-free materials such as polyethylene terephthalate glycol (PETG), which eliminate risks associated with di(2-ethylhexyl) phthalate (DEHP) leaching. Require ISO 10993-1 biocompatibility certification covering cytotoxicity, sensitization, and irritation.
Additionally, insist on extractables and leachables testing to confirm no harmful chemicals migrate into samples during storage—such contamination was linked to 17% of urine toxicology false positives in 2023. Manufacturers should provide batch-specific material safety data sheets (MSDS) and test reports to support material claims.
Sterility validation evidence: EO sterilization protocols and container integrity testing
Demand full ethylene oxide (EO) sterilization validation reports compliant with ISO 11135, including half-cycle studies, process development, and residual gas levels below 25 ppm. Accelerated aging data per ASTM F1980 must support the claimed shelf life.
Container integrity is equally vital. Validated test methods—such as dye ingress or vacuum decay—must demonstrate zero failure rates after simulated transport and storage. Sterility breaches in vacuum urine containers have led to recalls averaging $740,000 (Ponemon 2023), underscoring the importance of robust validation.
Key Red Flags in Supplier Documentation and Audit Readiness
Labeling and IFU Gaps: Missing UDI, Contraindications, or Single-Use Declarations
Incomplete labeling or Instructions for Use (IFU) pose significant regulatory risks. Missing UDI codes, contraindications for specific populations, or unclear single-use declarations violate FDA 21 CFR Part 801 and ISO 15223-1:2021 symbol requirements.
Facilities face a 33% higher audit failure rate when UDI data lacks machine-readable formatting or omits production identifiers like lot numbers and expiration dates. Ensure labels include UDI-DI and UDI-PI, lot number, expiry, and the single-use symbol to maintain compliance.
Common Audit Nonconformities: Inadequate Supplier Controls, DHF Gaps, and Sterility Revalidation Lapses
Audit failures frequently stem from three root causes: insufficient supplier qualification, incomplete Design History Files (DHF), and expired sterility revalidation beyond the FDA-recommended 24-month cycle. These issues account for 48% of ISO 13485:2016 nonconformities and are often linked to weak change control or undocumented corrective actions.
Manufacturers with unresolved DHF gaps experience 2.7 times more recalls. Prioritize suppliers with rigorous documentation practices, including traceable risk assessments, verification records, and timely revalidation protocols.
Making the Final Selection: A Practical Compliance Checklist for Buyers
Selecting compliant vacuum urine containers requires a structured evaluation. Use this checklist to ensure due diligence:
- Confirm explicit ISO 13485:2016 certification scope: The certificate must specifically list vacuum urine containers. Request documentation and verify its validity with the notified body.
- Verify FDA clearance pathway: Cross-check the 510(k) number in the FDA’s 510(k) Premarket Notification database to confirm it covers your intended model and use case.
- Demand material safety data: Review ISO 10993-1 biocompatibility reports and require extractables/leachables testing for PVC-free materials to ensure chemical stability.
- Inspect sterility validation: Require ISO 11135-compliant EO sterilization reports and container integrity testing (e.g., ASTM F3039 dye ingress). Revalidation should occur at least biannually or after any design change.
- Audit UDI and labeling: Confirm primary labels display UDI-DI/UDI-PI, lot number, expiry date, and the single-use symbol. IFUs must clearly state contraindications, such as restrictions for pediatric use.
- Assess supplier controls: Review audit history for recurring issues like inadequate supplier qualifications or DHF gaps. Favor manufacturers with no FDA Form 483 observations in the past 24 months.
- Evaluate total cost of compliance: Consider long-term costs including documentation management, training, and potential audit support. Non-compliant devices can trigger recalls exceeding $500,000 (FDA 2023), far outweighing initial savings.
FAQ Section
What is ISO 13485:2016 and why is it important for vacuum urine containers?
ISO 13485:2016 is a standard for quality management systems specific to medical devices, ensuring consistent design, development, and production. For vacuum urine containers, adherence to this standard guarantees safety, reliability, and compliance with international regulations.
How are vacuum urine containers classified by the FDA?
Vacuum urine containers are classified as FDA Class II devices, which require 510(k) clearance demonstrating substantial equivalence to an existing device.
What is UDI and why is it critical for compliance?
UDI stands for Unique Device Identification, a system that assigns distinct codes to medical devices to enhance traceability throughout the supply chain. Compliance with UDI is crucial for tracking issues and maintaining accountability.
What should a buyer check when choosing a vacuum urine container manufacturer?
Buyers should verify the manufacturer's ISO 13485:2016 certification scope, FDA 510(k) clearance, material safety data, sterility validation, UDI and labeling accuracy, and the supplier's audit readiness and history.